负载型催化剂由于具有稳定的界面结构、便于分离回收,被广泛用于石油化工、汽车尾气处理、新能源电池等领域。对负载型催化剂而言,金属纳米颗粒和载体之间存在不可避免的相互作用,包括两者之间的电荷转移,颗粒形貌和化学组成的改变,以及在载体迁移后形成包覆结构。这些界面相互作用共同影响了反应物的吸附和转化过程,是决定催化性能的关键因素。
在各种界面效应中,电子效应对催化性能的调节最为显著。界面电子结构与表面物种的吸附强弱密切相关,而表面物种的吸附直接影响了催化活性和选择性。研究发现,反应催化活性与物种吸附强度通常呈火山关系,只有适中的物种吸附强度才能到达最高的催化性能。然而,如何通过化学手段精准调控界面电子效应,使重要中间体吸附强度达到最适合的位置,是催化研究中的难点;同时,对于复杂反应何为重要中间体也不明确。因此,亟待开发精准的界面调控策略,进而解析复杂反应的构效关系,指导包括CO2转化、氢能再生等关键能源反应的催化材料设计。
最近,张亚文课题组从CeO2基负载型催化材料出发,开发了两种精准调控界面电子结构的方法,电化学诱导界面调控策略和氨热处理界面调控策略,分别实现了增强界面电子相互作用和减弱界面电子相互作用(图1)。
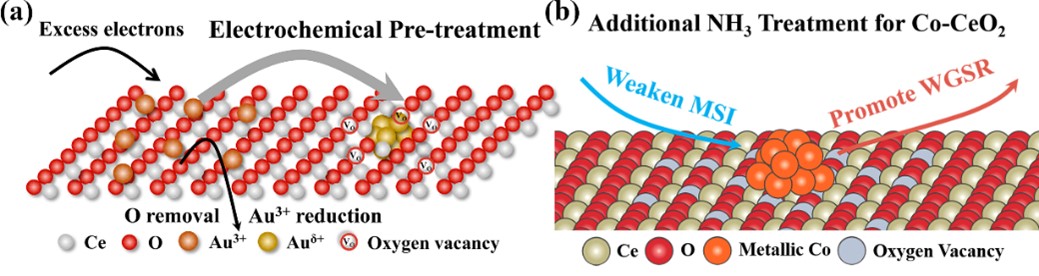
图1:调控策略示意图。(a) 电化学诱导界面调控策略;(b) 氨热处理界面调控策略。
电化学诱导策略如下:首先在CeO2表面负载Au(OH)3物种,在随后的电化学预处理过程中,利用Au3+物种的强氧化性,在其被还原的同时可以诱导CeO2载体的还原,进而增加两者的相互作用。得到的催化材料Au-CeO2-DP用在了CO2电催化还原反应中,在-0.7至-1.0 V的宽电位下均表现出95%以上的CO法拉第效率(图2)。得益于界面电子态的调控,-0.7 V的Au质量电流密度比传统NaBH4还原得到的催化剂提升了5.8倍,与文献中报道的结果比较也处在领先的水平。后续研究表明,增强的金属-载体相互作用使Au纳米颗粒表现出δ+的价态,同时在界面处产生了丰富的氧空位。界面电子结构的改变提升了催化材料对CO2的吸附稳定性,加速了重要羧酸中间体生成,进而提升CO2电还原反应的催化性能。
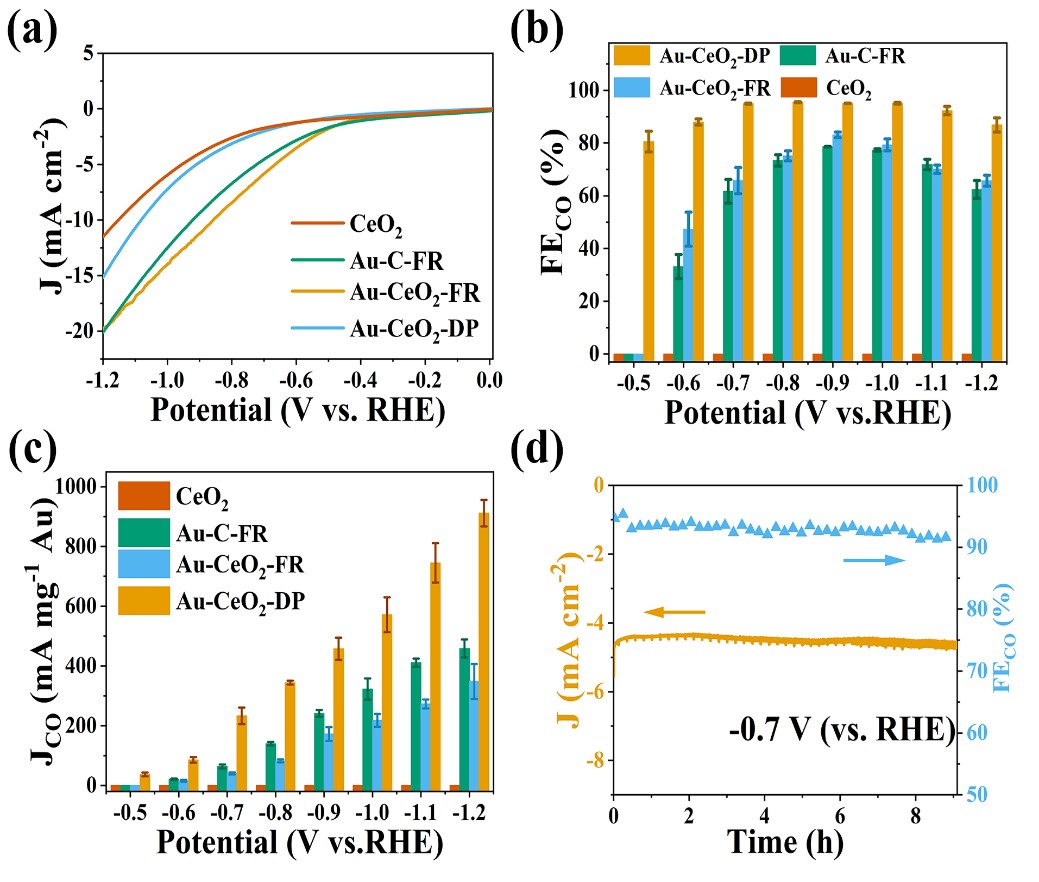
图2:Au-CeO2-DP的催化性能。(a) LSV曲线;(b) CO2电催化还原活性;(c) Au质量电流密度;(d) 催化稳定性。
氨热处理策略通过对CeO2纳米结构的NH3热处理过程,在CeO2中引入N掺杂,从而对氧空位进行封闭,减弱其与表面金属物种的相互作用。通过这种合成策略得到的Co-CeO2催化剂用于水煤气变换制氢反应,发现随着NH3处理温度的升高,催化活性逐渐增强,其中800度处理的样品Co/800N-CeO2催化活性是未经处理样品的23.8倍,与文献结果比较表明,减弱的界面电子相互作用大幅提升了催化效率,使Co基催化剂用于工业生产中成为可能。活性的提高主要来自两个方面,一方面,由于金属-载体相互作用减弱,Co物种在反应条件下平均价态降低,0价Co位点的增加有利于稳定CO吸附,并加速重要中间体羧酸盐的生成;另一方面,N物种在反应条件下不稳定,N物种的离去有利于氧空位的生成,从而增强对水分子的活化能力。两者共同作用提高水煤汽变换反应的催化性能。

图3:Co-CeO2催化材料的水煤汽变换反应催化性能。(a) CO转化率随温度的变化曲线;(b) 280 ℃的反应速率。
对金属-载体相互作用的两部分研究工作“Au3+ Species-Induced Interfacial Activation Enhances Metal−Support Interactions for Boosting Electrocatalytic CO2 Reduction to CO”和Weakening the Metal−Support Interactions of M/CeO2 (M = Co, Fe, Ni) Using a NH3‑Treated CeO2 Support for an Enhanced Water−Gas Shift Reaction,近期在ACS Catal.上发表(ACS Catal. 2022, 12, 923−934; ACS Catal. 2022, 12, 11942−11954)。第一作者为张亚文课题组博士生孙啸尘,张亚文教授为该工作的通讯作者。刘海超教授课题组也对该研究做出了实质贡献。这一系列工作为开发稳定、高活性负载型催化剂提供了新的思路。
该研究得到国家自然科学基金委重点项目、国家重点研发计划和北京分子科学国家研究中心的资助,还得到严纯华院士与孙聆东教授课题组的大力支持。
原文链接:https://doi.org/10.1021/acscatal.1c05503
https://doi.org/10.1021/acscatal.2c03664

 北大化学微信
北大化学微信