继2021年9月联合华为云发布蛋白质多序列比对开源数据集后,北京大学化学与分子工程学院、北京大学生物医学前沿创新中心(BIOPIC)、昌平实验室、深圳湾实验室高毅勤课题组在蛋白质结构预测领域再次取得突破性进展。近日,他们联合华为昇思MindSpore团队推出了基于AlphaFold2算法的蛋白质结构预测推理工具,相关模型代码依托于华为全场景AI计算框架MindSpore进行开源开放、定期扩展与维护,旨在为全世界相关的产、学、研团队提供优质的国产软硬件解决方案。
蛋白质在分子生物学的中心法则中具有重要地位,在各种生命过程中不可或缺,但是传统的蛋白质结构解析实验难度大,而计算机预测方法则一直存在计算精度不足的缺陷。2020年谷歌DeepMind团队发布了人工智能蛋白质结构预测模型AlphaFold2【1】,使得计算机蛋白质结构预测工具精度可与实验方法相媲美,被Nature等杂志喻为“前所未有的进步”。本次高毅勤课题组推出的蛋白结构预测工具依托华为昇思MindSpore,可对氨基酸序列长度2000+的蛋白质结构进行解析,能覆盖约99%以上的蛋白序列【2】。同时,借助异构计算架构CANN(Compute Architecture for Neural Network)释放昇腾AI处理器的澎湃算力,通过软硬件协同优化大大提高了蛋白质预测的计算效率。参考DeepMind于2021年7月份开源的蛋白质结构预测模型AlphaFold2【3】,本次开源的蛋白质结构预测推理工具模型部分与其相同,在多序列比对阶段则采用了MMseqs2进行序列检索【4】,相比于原版算法端到端运算速度有2-3倍的提升。准确、快速的蛋白质结构预测不仅可以在学术研究领域帮助科学家快速获得或验证关键蛋白结构,从而促进人们对重要生命过程分子机理的理解,而且在医疗健康和生物工程领域也将作为有力的工具,引发药物靶点发现、功能蛋白设计(如抗体和人工酶)以及大分子(蛋白)或小分子药物高通量虚拟筛选的革命。
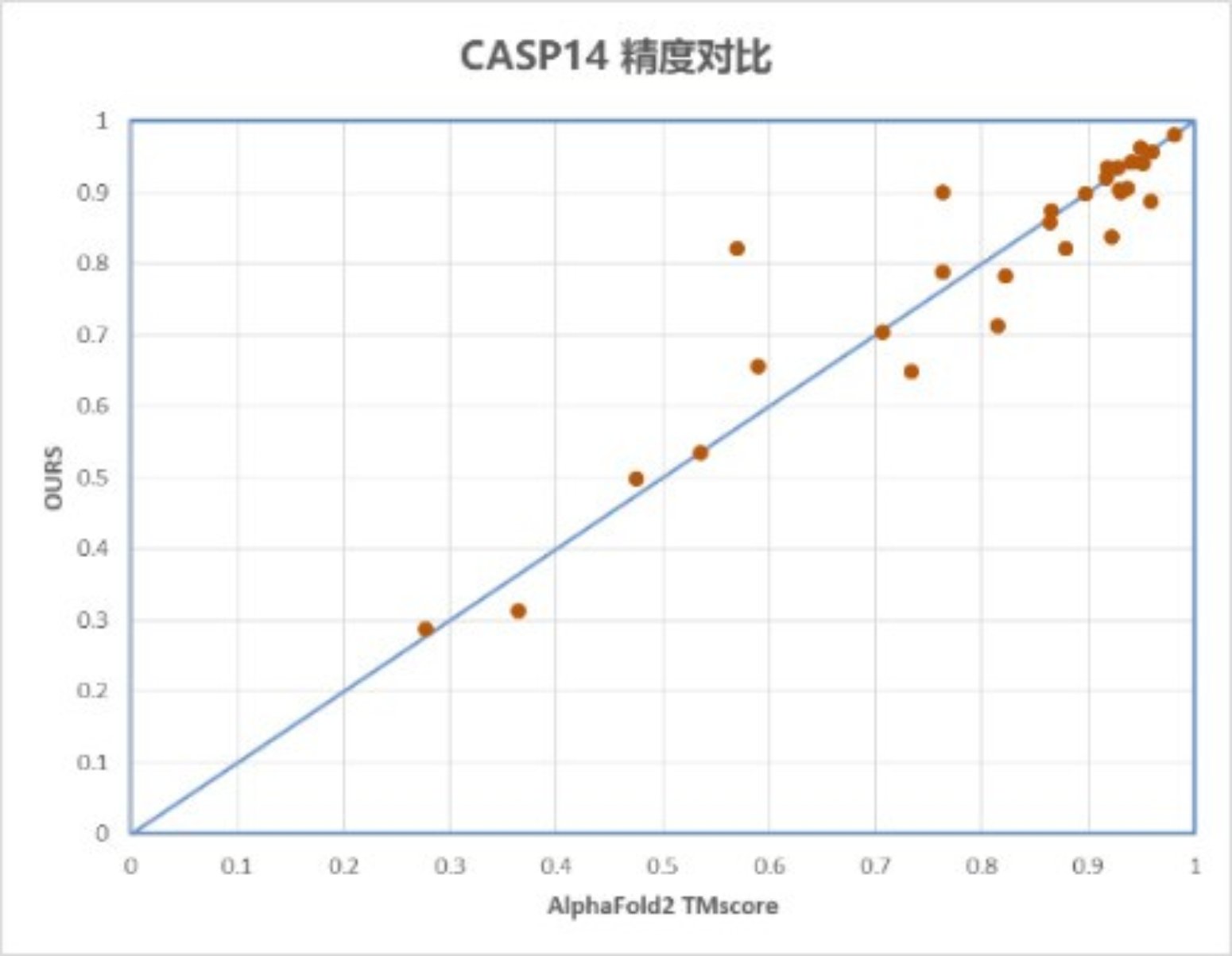
图1 昇思MindSpore模型与AlphaFold2精度对比

图2 通过昇思MindSpore预测的T1079(绿色代表实验结构,红色代表预测结构)
针对蛋白质结构预测及折叠问题,联合团队未来还会发布创新的全栈(算法+软件+硬件)国产自主可控技术,同时也会牵手更多的学术科研界合作伙伴,在计算生物、计算材料、工业仿真等更广泛的科学计算领域深耕,促进国内相关基础研究的发展。
【1】Jumper J, Evans R, Pritzel A, et al. Applying and improving AlphaFold at CASP14[J]. Proteins: Structure, Function, and Bioinformatics, 2021.
【2】https://ftp.uniprot.org/pub/databases/uniprot/previous_releases/release-2021_02/
knowledgebase/UniProtKB_TrEMBL-relstat.html
【3】Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold[J]. Nature, 2021, 596(7873): 583-589.
【4】Mirdita M, Ovchinnikov S, Steinegger M. ColabFold-Making protein folding accessible to all[J]. BioRxiv, 2021.
代码开源路径:
https://gitee.com/mindspore/mindscience/tree/master/MindSPONGE/mindsponge/fold

 北大化学微信
北大化学微信