自从二苯铬发现以来,金属芳烃化合物一直是金属有机化学研究的重要领域。首例反三明治型甲苯桥连的双铀配合物于2000年被报道(图1左)。随后,多个课题组相继报道了不同配体支撑的反三明治型芳烃桥联的双铀配合物。对其电子结构与成键性质的研究表明,铀通过5f与6d轨道与芳烃的π*轨道形成δ键。此外,这类铀-芳烃相互作用在铀配合物的电催化分解水过程中也起了关键的“蓄电池”作用(图1中)。尽管有较多反三明治型芳烃桥联的双铀配合物以及结构相似的稀土金属配合物的报道,但是迄今尚没有钍的类似物的合成。近日,北京大学化学与分子工程学院黄闻亮课题组与美国加州大学洛杉矶分校Paula Diaconescu课题组合作,报道了首例反三明治型芳烃桥连的双钍配合物的合成与表征,并对其进行了反应性和电子结构的研究。该成果以“Arene-Bridged Dithorium Complexes: Inverse Sandwiches Supported by a δ Bonding Interaction”为题,在《美国化学会志》(Journal of the American Chemical Society)上发表。
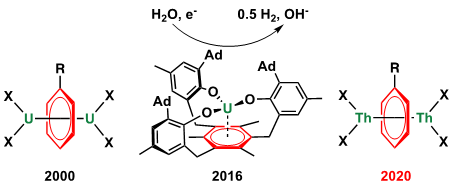
图1:反三明治型锕系金属芳烃配合物与铀-芳烃相互作用在催化中的作用。
四价钍前驱体可由二茂铁二胺配体钾盐与四氯化钍通过盐类复分解反应制备(图2上a)。在石墨钾(KC8)作用下,四价钍前驱体可与苯、甲苯、联苯或萘等反应得到一系列反三明治型芳烃桥连的双钍配合物Th2-arene(图2上b-e)。这些钍的芳烃配合物之间可以发生芳烃交换反应,根据其反应性可以得到Th2-arene的相对热力学稳定性:Th2-naph < Th2-tol < Th2-benzene ≈ Th2-biph。单晶X射线衍射揭示了这些双钍配合物中芳烃都采用μ-η6,η6-的配位模式(图2下),其中配位的芳环中的碳–碳键键长明显长于中性的芳环。特别地,在Th2-naph中,非配位的芳环上的碳–碳键键长呈现单双键交替的现象,表明非配位芳环出现了去芳构化,这可能是Th2-naph稳定性较低的原因。单晶结构数据与波谱学表征(核磁共振波谱、紫外-可见-近红外吸收光谱、X射线吸收谱等)均表明,在Th2-arene中,钍为正四价,而配位的芳环为具有休克尔芳香性的负四价阴离子。
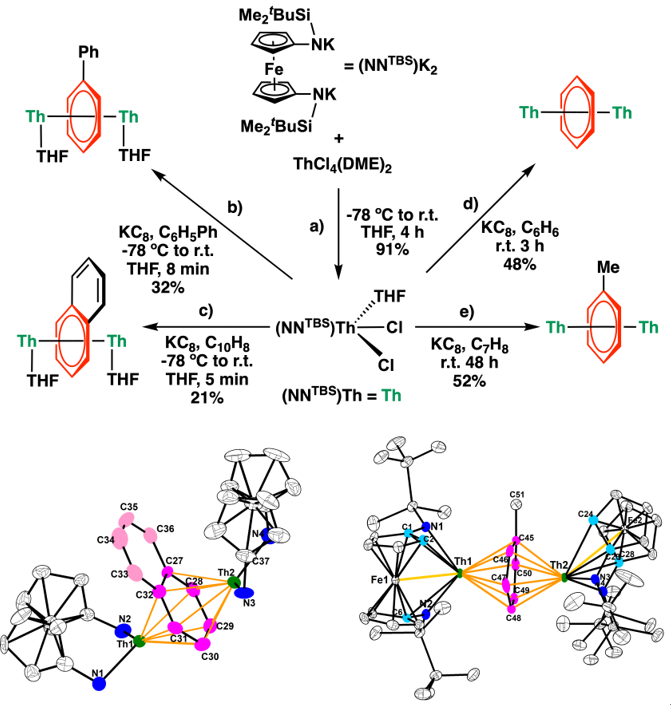
图2:Th2-arene的合成(上)与Th2-tol(下左)和Th2-naph(下右)的结构。
反应性研究表明,Th2-arene可以还原环辛四烯、偶氮苯、二苯乙炔等不饱和小分子(图3左),得到相应的单核钍配合物。其中,Th2-naph表现出最高的反应活性,其不仅可以还原大空阻的双(三甲基硅基)乙炔得到钍杂环丙烷产物,还能与蒽反应得到单核钍的蒽配合物。值得注意的是,单核钍的蒽配合物能够还原苯得到Th2-benzene,这是首个金属蒽化合物还原苯的例子。
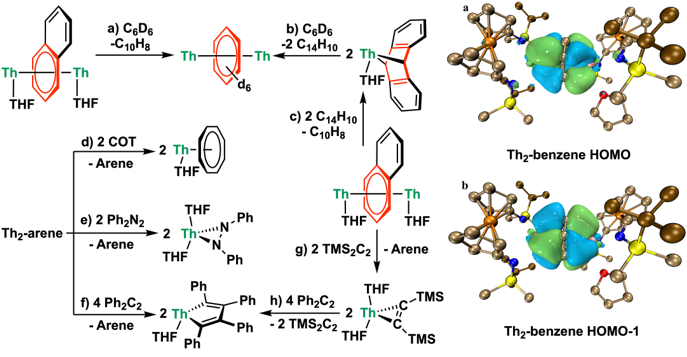
图3:Th2-arene的反应性(左)与电子结构(右)。
为了加深对反三明治型芳烃桥联双钍配合物的电子结构与成键性质的理解,研究者利用相对论校正的密度泛函理论对其进行了分析。计算得到的优化结构与单晶结构数据高度吻合。图3右侧展示了Th2-benzene的HOMO和HOMO-1,二者在能量上几乎简并(-4.04 eV、-4.07 eV)。两者均为具有δ对称性的成键轨道,分别由配位苯环的π4或π5轨道与钍的6dδ和5fδ轨道组成。轨道成分分析表明,在HOMO中,苯上碳的2p轨道的贡献率为66.5%,而钍的轨道的贡献率为28.2% (6d: 15.8%; 5f: 12.4%);在HOMO-1中,苯上碳的2p轨道的贡献率为67.4%,而钍的轨道的贡献率为28.7% (6d: 16.9%; 5f: 11.8%)。对其他Th2-arene的计算分析也表明,这种共价性的δ相互作用对于稳定Th2-arene的反三明治型结构起到了至关重要的作用。此外,与结构类似的反三明治型芳烃桥联双稀土金属或双铀配合物的比较显示,钍与芳烃成键的共价性(约30%)介于稀土金属(钇:约20%)与铀(约50%)之间。
综上,这项工作首次合成并表征了反三明治型芳烃(苯、甲苯、联苯、萘)桥连的双钍配合物。钍的芳烃配合物之间的交换反应揭示了它们的热力学稳定性差异,而初步的反应性研究表明这类钍的芳烃配合物可以进行多电子的氧化还原反应。理论计算分析阐明了其电子结构与成键性质,揭示了反三明治型芳烃桥联的双钍配合物中存在具有一定共价性的δ键。北京大学化学与分子工程学院的BMS Junior Fellow 于超博士为本论文第一作者,北京大学黄闻亮特聘研究员、美国加州大学洛杉矶分校Paula Diaconescu教授为共同通讯作者。本研究得到了北京大学、北京分子科学国家研究中心等的经费支持。
原文链接:https://dx.doi.org/10.1021/jacs.0c11215

 北大化学微信
北大化学微信