
2013 - present

Abstract
Optical biosensors have been invaluable tools in neuroscience research, as they provide the ability to directly visualize neural activity in real time, with high specificity, and with exceptional spatial and temporal resolution. Notably, a majority of these sensors are based on fluorescent protein scaffolds, which offer the ability to target specific cell types or even subcellular compartments. However, fluorescent proteins are intrinsically bulky tags, often insensitive to the environment, and always require excitation light illumination. To address these limitations, there has been a proliferation of alternative sensor scaffolds developed in recent years, including hybrid sensors that combine the advantages of synthetic fluorophores and genetically encoded protein tags, as well as bioluminescent probes. While still in their early stage of development as compared with fluorescent protein-based sensors, these novel probes have offered complementary solutions to interrogate various aspects of neuronal communication, including transmitter release, changes in membrane potential, and the production of second messengers. In this Review, we discuss these important new developments with a particular focus on design strategies.
A. Wang, J. Feng, Y. Li* & P. Zou*.(2018). ACS. Chem. Neurosci., (in press)

Membrane voltage is an important biophysical signal that underlies intercellular electrical communications. In this study, we present a novel fluorescent voltage indicator that enables the investigation of electrical signaling at high spatial resolution. Our method is built upon the site-specific modification of microbial rhodopsin proteins with organic fluorophores, resulting in a hybrid indicator scaffold that represents one of the most sensitive and fastest orange-colored voltage indicators developed to date. We applied this technique to optically map electrical connectivity in cultured cells, which revealed gap junction-mediated long-range coupling that spanned over hundreds of micrometers.
Y. Xu†, L. Peng†, S. Wang†, A. Wang†, R. Ma, Y. Zhou, J. Yang, D. E. Sun, W. Lin, X. Chen, P. Zou*.(2018). Angew. Chem. Int. Ed. Engl., 57, 3949-3553.
Abstract
We developed membrane voltage nanosensors that are based on inorganic semiconductor nanoparticles. We provide here a feasibility study for their utilization. We use a rationally designed peptide to functionalize the nanosensors, imparting them with the ability to self-insert into a lipid membrane with a desired orientation. Once inserted, these nanosensors could sense membrane potential via the quantum confined Stark effect, with a single-particle sensitivity. With further improvements, these nanosensors could potentially be used for simultaneous recording of action potentials from multiple neurons in a large field of view over a long duration and for recording electrical signals on the nanoscale, such as across one synapse.
K. Park, Y. Kuo, V. Shvadchak, A. Ingargiola, X. Dai, L. Hsiung, W. Kim, H. Zhou, P. Zou, A. J. Levine, J. Li, S. Weiss. (2018). Sci. Adv., 4, e1601453.
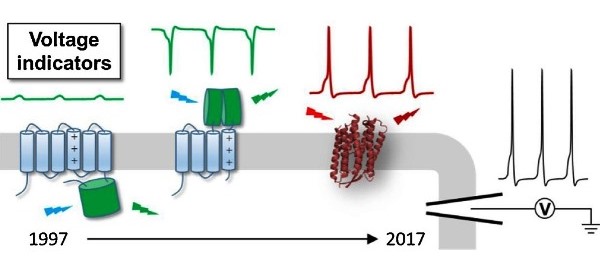
Abstract
A holy grail in neuroscience is to understand how brain functions arise from neural network-level electrical activities. Voltage imaging allows for the direct visualization of electrical signaling at high spatial and temporal resolutions across a large neuronal population. Central to this technique is a palette of genetically-encoded fluorescent probes with fast and sensitive voltage responses. In this review, we chronicle the development and applications of genetically-encoded voltage indicators (GEVIs) over the past two decades, with a primary focus on the structural design that harness the power of fluctuating transmembrane electric fields. We hope this article will inform chemical biologists and protein engineers of the GEVI history and inspire novel design ideas.
L. Peng†, Y. Xu†, and P. Zou (2017). Chin. Chem. Lett., 28, 1925-1928.
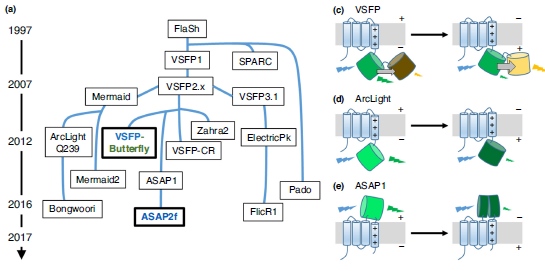

Abstract
Membrane voltages are ubiquitous throughout cell biology. Voltage is most commonly associated with excitable cells such as neurons and cardiomyocytes, although many other cell types and organelles also support electrical signaling. Voltage imaging in vivo would offer unique capabilities in reporting the spatial pattern and temporal dynamics of electrical signaling at the cellular and circuit levels. Voltage is not directly visible, and so a longstanding challenge has been to develop genetically encoded fluorescent voltage indicator proteins. Recent advances have led to a profusion of new voltage indicators, based on different scaffolds and with different tradeoffs between voltage sensitivity, speed, brightness, and spectrum. In this review, we describe recent advances in design and applications of genetically-encoded voltage indicators (GEVIs). We also highlight the protein engineering strategies employed to improve the dynamic range and kinetics of GEVIs and opportunities for future advances.
Y. Xu, P. Zou#, A. E. Cohen# (2017). Curr. Opin. Chem. Biol., 39, 1-10.
Optical imaging of voltage indicators based on green fluorescent proteins (FPs) or archaerhodopsin has emerged as a powerful approach for detecting the activity of many individual neurons with high spatial and temporal resolution. Relative to green FP-based voltage indicators, a bright red-shifted FP-based voltage indicator has the intrinsic advantages of lower phototoxicity, lower autofluorescent background, and compatibility with blue-light-excitable channelrhodopsins. Here, we report a bright red fluorescent voltage indicator (fluorescent indicator for voltage imaging red; FlicR1) with properties that are comparable to the best available green indicators. To develop FlicR1, we used directed protein evolution and rational engineering to screen libraries of thousands of variants. FlicR1 faithfully reports single action potentials (∼3% ΔF/F) and tracks electrically driven voltage oscillations at 100 Hz in dissociated Sprague Dawley rat hippocampal neurons in single trial recordings. Furthermore, FlicR1 can be easily imaged with wide-field fluorescence microscopy. We demonstrate that FlicR1 can be used in conjunction with a blue-shifted channelrhodopsin for all-optical electrophysiology, although blue light photoactivation of the FlicR1 chromophore presents a challenge for applications that require spatially overlapping yellow and blue excitation.
A. S. Abdelfattah, S. L. Farhi, Y. Zhao, D. Brinks, P. Zou, A. Ruangkittisakul, J. Platisa, V. A. Pieribone, K. Ballanyi, A. E. Cohen, R. E. Campbell. (2016). J Neurosci., 36, 2458-2472.
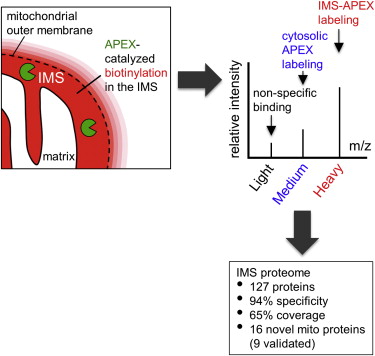
Abstract
Obtaining complete protein inventories for subcellular regions is a challenge that often limits our understanding of cellular function, especially for regions that are impossible to purify and are therefore inaccessible to traditional proteomic analysis. We recently developed a method to map proteomes in living cells with an engineered peroxidase (APEX) that bypasses the need for organellar purification when applied to membrane-bound compartments; however, it was insufficiently specific when applied to unbounded regions that allow APEX-generated radicals to escape. Here, we combine APEX technology with a SILAC-based ratiometric tagging strategy to substantially reduce unwanted background and achieve nanometer spatial resolution. This is applied to map the proteome of the mitochondrial intermembrane space (IMS), which can freely exchange small molecules with the cytosol. Our IMS proteome of 127 proteins has >94% specificity and includes nine newly discovered mitochondrial proteins. This approach will enable scientists to map proteomes of cellular regions that were previously inaccessible.
V. Hung, P. Zou†, H. Rhee†, N. D. Udeshi, V. Cracan, T. Svinkina, S. A. Carr, V. K. Mootha, and A. Y. Ting. (2014). Mol. Cell, 55, 332-341. († equal contribution)
Genetically encoded fluorescent reporters of membrane potential promise to reveal aspects of neural function not detectable by other means. We present a palette of multicoloured brightly fluorescent genetically encoded voltage indicators with sensitivities from 8–13% ΔF/F per 100 mV, and half-maximal response times from 4–7 ms. A fluorescent protein is fused to an archaerhodopsin-derived voltage sensor. Voltage-induced shifts in the absorption spectrum of the rhodopsin lead to voltage-dependent nonradiative quenching of the appended fluorescent protein. Through a library screen, we identify linkers and fluorescent protein combinations that report neuronal action potentials in cultured rat hippocampal neurons with a single-trial signal-to-noise ratio from 7 to 9 in a 1 kHz imaging bandwidth at modest illumination intensity. The freedom to choose a voltage indicator from an array of colours facilitates multicolour voltage imaging, as well as combination with other optical reporters and optogenetic actuators.
P. Zou†, Y. Zhao†, A. D. Douglass, D. R. Hochbaum, D. Brinks, C. A. Werley, D. J. Harrison, R. E. Campbell, and A. E. Cohen. (2014). Nat. Commun., 5, 4625. († equal contribution)
DOI: 10.1038/ncomms5625
All-optical electrophysiology—spatially resolved simultaneous optical perturbation and measurement of membrane voltage—would open new vistas in neuroscience research. We evolved two archaerhodopsin-based voltage indicators, QuasAr1 and QuasAr2, which show improved brightness and voltage sensitivity, have microsecond response times and produce no photocurrent. We engineered a channelrhodopsin actuator, CheRiff, which shows high light sensitivity and rapid kinetics and is spectrally orthogonal to the QuasArs. A coexpression vector, Optopatch, enabled cross-talk–free genetically targeted all-optical electrophysiology. In cultured rat neurons, we combined Optopatch with patterned optical excitation to probe back-propagating action potentials (APs) in dendritic spines, synaptic transmission, subcellular microsecond-timescale details of AP propagation, and simultaneous firing of many neurons in a network. Optopatch measurements revealed homeostatic tuning of intrinsic excitability in human stem cell–derived neurons. In rat brain slices, Optopatch induced and reported APs and subthreshold events with high signal-to-noise ratios. The Optopatch platform enables high-throughput, spatially resolved electrophysiology without the use of conventional electrodes.
D. R. Hochbaum, Y. Zhao, S. Farhi, N. Klapoetke, C. A. Werley, V. Kapoor, P. Zou, J. M. Kralj, D. Maclaurin, N. Smedemark-Margulies, J. Saulnier, G. Boulting, Y. Cho, M. Melkonian, G. K. -. Wong, D. J. Harrison, V. N. Murthy, B. Sabatini, E. S. Boyden, R. E. Campbell, and A. E. Cohen. (2014). Nat. Methods, 11, 825-833.
DOI: 10.1038/nmeth.3000
The low-density lipoprotein receptor (LDLR) is a critical determinant of plasma cholesterol levels that internalizes lipoprotein cargo via clathrin-mediated endocytosis. Here, we show that the E3 ubiquitin ligase IDOL stimulates a previously unrecognized, clathrin-independent pathway for LDLR internalization. Real-time single-particle tracking and electron microscopy reveal that IDOL is recruited to the plasma membrane by LDLR, promotes LDLR internalization in the absence of clathrin or caveolae, and facilitates LDLR degradation by shuttling it into the multivesicular body (MVB) protein-sorting pathway. The IDOL-dependent degradation pathway is distinct from that mediated by PCSK9 as only IDOL employs ESCRT (endosomal-sorting complex required for transport) complexes to recognize and traffic LDLR to lysosomes. Small interfering RNA (siRNA)-mediated knockdown of ESCRT-0 (HGS) or ESCRT-I (TSG101) components prevents IDOL-mediated LDLR degradation. We further show that USP8 acts downstream of IDOL to deubiquitinate LDLR and that USP8 is required for LDLR entry into the MVB pathway. These results provide key mechanistic insights into an evolutionarily conserved pathway for the control of lipoprotein receptor expression and cellular lipid uptake.
E. Scotti, M. Calamai, C. N. Goulbourne, L. Zhang, C. Hong, R. R. Lin, J. Choi, P. F. Pilch, L. G. Fong, P. Zou, A. Y. Ting, F. S. Pavone, S. G. Young, and P. Tontonoz. (2013). Mol. Cell. Biol., 33, 1503-1514.
DOI: 10.1128/MCB.01716-12
Microscopy and mass spectrometry (MS) are complementary techniques: The former provides spatiotemporal information in living cells, but only for a handful of recombinant proteins at a time, whereas the latter can detect thousands of endogenous proteins simultaneously, but only in lysed samples. Here, we introduce technology that combines these strengths by offering spatially and temporally resolved proteomic maps of endogenous proteins within living cells. Our method relies on a genetically targetable peroxidase enzyme that biotinylates nearby proteins, which are subsequently purified and identified by MS. We used this approach to identify 495 proteins within the human mitochondrial matrix, including 31 not previously linked to mitochondria. The labeling was exceptionally specific and distinguished between inner membrane proteins facing the matrix versus the intermembrane space (IMS). Several proteins previously thought to reside in the IMS or outer membrane, including protoporphyrinogen oxidase, were reassigned to the matrix by our proteomic data and confirmed by electron microscopy. The specificity of peroxidase-mediated proteomic mapping in live cells, combined with its ease of use, offers biologists a powerful tool for understanding the molecular composition of living cells.
H. W. Rhee†, P. Zou†, N. D. Udeshi, J. D. Martell, V. K. Mootha, S. A. Carr, and A. Y. Ting. (2013). Science, 339, 1328-1331. († equal contribution)
2008-2012
A screen of Trp37 mutants of Escherichia coli lipoic acid ligase (LplA) revealed enzymes capable of ligating an aryl-aldehyde or aryl-hydrazine substrate to LplA's 13-residue acceptor peptide. Once site-specifically attached to recombinant proteins fused to this peptide, aryl-aldehydes could be chemoselectively derivatized with hydrazine-probe conjugates, and aryl-hydrazines could be derivatized in an analogous manner with aldehyde-probe conjugates. Such two-step labeling was demonstrated for AlexaFluor568 targeting to monovalent streptavidin in vitro, and to neurexin-1β on the surface of living mammalian cells. To further highlight this technique, we labeled the low-density lipoprotein receptor on the surface of live cells with fluorescent phycoerythrin protein to allow single-molecule imaging and tracking over time.
J. D. Cohen, P. Zou, and A. Y. Ting. (2012). Chembiochem, 13, 888-894.
05. Foldon unfolding mediates the interconversion between Mpro-C monomer and 3D domain-swapped dimer
The C-terminal domain (Mpro-C) of SARS-CoV main protease adopts two different fold topologies, a monomer and a 3D domain-swapped dimer. Here, we report that Mpro-C can reversibly interconvert between these two topological states under physiological conditions. Although the swapped α1-helix is fully buried inside the protein hydrophobic core, the interconversion of Mpro-C is carried out without the hydrophobic core being exposed to solvent. The 3D domain swapping of Mpro-C is activated by an order-to-disorder transition of its C-terminal α5-helix foldon. Unfolding of this foldon promotes self-association of Mpro-C monomers and functions to mediate the 3D domain swapping, without which Mpro-C can no longer form the domain-swapped dimer. Taken together, we propose that there exists a special dimeric intermediate enabling the protein core to unpack and the α1-helices to swap in a hydrophobic environment, which minimizes the energy cost of the 3D domain-swapping process.
J. D. Cohen, P. Zou, and A. Y. Ting. (2012). Chembiochem, 13, 888-894.
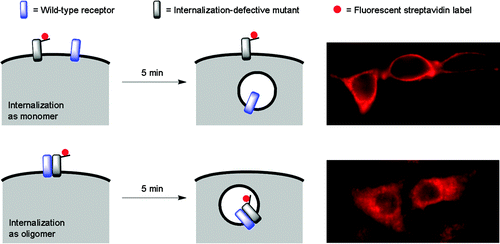
Abstract
Methods to probe receptor oligomerization are useful to understand the molecular mechanisms of receptor signaling. Here we report a fluorescence imaging method to determine receptor oligomerization state in living cells during endocytic internalization. The wild-type receptor is co-expressed with an internalization-defective mutant, and the internalization kinetics of each are independently monitored. If the receptor internalizes as an oligomer, then the wild-type and mutant isoforms will mutually influence each others’ trafficking properties, causing co-internalization of the mutant or co-retention of the wild-type at the cell surface. Using this approach, we found that the low density lipoprotein (LDL) receptor internalizes as an oligomer into cells, both in the presence and absence of LDL ligand. The internalization kinetics of the wild-type receptor are not changed by LDL binding. We also found that the oligomerization domain of the LDL receptor is located in its cytoplasmic tail.
P. Zou, and A. Y. Ting. (2011). ACS Chem. Biol., 6, 308-313.
DOI: 10.1021/cb100361k
Heparin-binding EGF-like growth factor (HB-EGF) is a ligand for EGF receptor (EGFR) and possesses the ability to signal in juxtacrine, autocrine and/or paracrine mode, with these alternatives being governed by the degree of proteolytic release of the ligand. Although the spatial range of diffusion of released HB-EGF is restricted by binding heparan-sulfate proteoglycans (HSPGs) in the extracellular matrix and/or cellular glycocalyx, ascertaining mechanisms governing non-released HB-EGF localization is also important for understanding its effects. We have employed a new method for independently tracking the localization of the extracellular EGF-like domain of HB-EGF and the cytoplasmic C-terminus. A striking observation was the absence of the HB-EGF transmembrane pro-form from the leading edge of COS-7 cells in a wound-closure assay; instead, this protein localized in regions of cell-cell contact. A battery of detailed experiments found that this localization derives from a trans interaction between extracellular HSPGs and the HB-EGF heparin-binding domain, and that disruption of this interaction leads to increased release of soluble ligand and a switch in cell phenotype from juxtacrine-induced growth inhibition to autocrine-induced proliferation. Our results indicate that extracellular HSPGs serve to sequester the transmembrane pro-form of HB-EGF at the point of cell-cell contact, and that this plays a role in governing the balance between juxtacrine versus autocrine and paracrine signaling.
R. N. Prince, E. R. Schreiter, P. Zou, H. S. Wiley, A. Y. Ting, R. T. Lee, and D. A. Lauffenburger. (2010). J. Cell. Sci., 123, 2308-2318.
DOI: 10.1242/jcs.058321
SARS coronavirus main protease (Mpro) plays an essential role in the extensive proteolytic processing of the viral polyproteins (pp1a and pp1ab), and it is an important target for anti-SARS drug development. We have reported that both the Mpro C-terminal domain alone (Mpro-C) and the N-finger deletion mutant of Mpro(Mpro-Δ7) exist as a stable dimer and a stable monomer (Zhong et al., J Virol 2008; 82:4227-4234). Here, we report structures of both Mpro-C monomer and dimer. The structure of the Mpro-C monomer is almost identical to that of the C-terminal domain in the crystal structure of Mpro. Interestingly, the Mpro-C dimer structure is characterized by 3D domain-swapping, in which the first helices of the two protomers are interchanged and each is enwrapped by four other helices from the other protomer. Each folding subunit of the Mpro-C domain-swapped dimer still has the same general fold as that of the Mpro-C monomer. This special dimerization elucidates the structural basis for the observation that there is no exchange between monomeric and dimeric forms of Mpro-C and Mpro-Δ7.
N. Zhong, S. Zhang, F. Xue, X. Kang, P. Zou, J. Chen, C. Liang, Z. Rao, C. Jin, Z. Lou, and B. Xia. (2009). Protein Sci., 18, 839-844.
DOI: 10.1002/pro.76
The main protease (Mpro) of severe acute respiratory syndrome coronavirus (SARS-CoV) plays an essential role in the extensive proteolytic processing of the viral polyproteins (pp1a and pp1ab), and it is an important target for anti-SARS drug development. It was found that SARS-CoV Mpro exists in solution as an equilibrium of both monomeric and dimeric forms, and the dimeric form is the enzymatically active form. However, the mechanism of SARS-CoV Mpro dimerization, especially the roles of its N-terminal seven residues (N-finger) and its unique C-terminal domain in the dimerization, remain unclear. Here we report that the SARS-CoV Mpro C-terminal domain alone (residues 187 to 306; Mpro-C) is produced in Escherichia coli in both monomeric and dimeric forms, and no exchange could be observed between them at room temperature. The Mpro-C dimer has a novel dimerization interface. Meanwhile, the N-finger deletion mutant of SARS-CoV Mpro also exists as both a stable monomer and a stable dimer, and the dimer is formed through the same C-terminal-domain interaction as that in the Mpro-C dimer. However, no C-terminal domain-mediated dimerization form can be detected for wild-type SARS-CoV Mpro. Our study results help to clarify previously published controversial claims about the role of the N-finger in SARS-CoV Mpro dimerization. Apparently, without the N-finger, SARS-CoV Mpro can no longer retain the active dimer structure; instead, it can form a new type of dimer which is inactive. Therefore, the N-finger of SARS-CoV Mpro is not only critical for its dimerization but also essential for the enzyme to form the enzymatically active dimer.
N. Zhong, S. Zhang, P. Zou, J. Chen, X. Kang, Z. Li, C. Liang, C. Jin, and B. Xia. (2008). J. Virol., 82, 4227-4234.
DOI: 10.1128/JVI.02612-07




