双管齐下:北京大学化学院吕华课题组结合大环化和聚氨基酸偶联两种策略极大改善蛋白质体内药学活性
蛋白质是一类重要的临床药物,然而其体内药学表现受到了稳定性差,体内半衰期短,自身免疫源性高等缺陷的限制。对多肽/蛋白进行环化是一种增强多肽/蛋白体外活性和稳定性的常见手段;此外,环状多肽因其受限的构型和较小的分子尺寸,相较于线性多肽具有更好的组织渗透性。然而,由于缺乏必要的“隐身” 机制,环化多肽和蛋白的体内循环时间较短,研究大多停留于细胞层面,鲜有系统的活体药学研究报道。高分子偶联是一种延长蛋白质药物体内循环时间的常见方法。然而由于其较大的空间位阻效应,蛋白质-高分子偶联物的蛋白活性往往极大降低,并且组织渗透性较差。吕华课题组通过结合环化和高分子偶联两种方法,制备了蛋白质-聚氨基酸大环偶联物,获得了优异的体内药学效果(图1)。
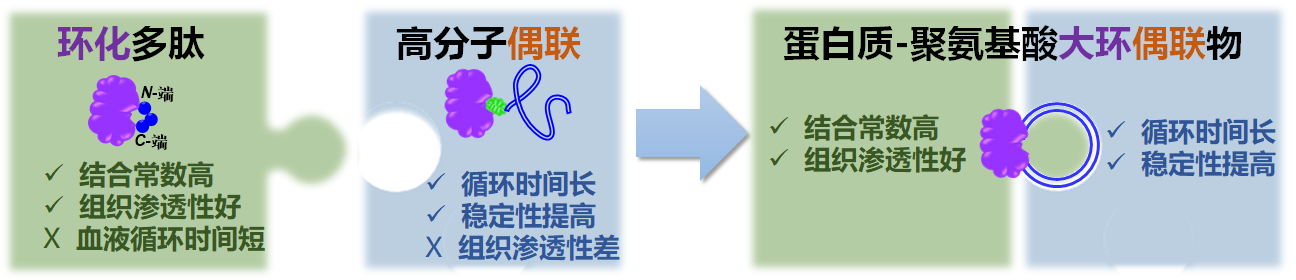
图1. 蛋白质-聚氨基酸大分子偶联物可同时具备环化以及高分子偶联两种策略的优点,从而体现出更优异的药学活性。
基于该实验室所发展的位点特异蛋白质-聚氨基酸偶联技术(J. Am. Chem. Soc. 2016, 138, 10995−11000),研究人员以干扰素-α2b(一种抗病毒和抗肿瘤药物)为模型药物蛋白合成了头-尾相接的干扰素-聚氨基酸大环偶联物,并将其与野生型干扰素,2个线型干扰素-聚氨基酸偶联物,以及1个线型干扰素-PEG偶联物平行比较(图2)。研究结果表明,无论在细胞实验还是动物实验层面,干扰素-聚氨基酸大环偶联物的药学性质均明显优于其他对照组。最为特殊的是,大环偶联物不仅仅具有传统蛋白质-高分子偶联物的典型优势如长循环时间和高肿瘤滞留,还有环状多肽药物特有的高肿瘤渗透性(图2)。由于这一系列的优异性质,干扰素-聚氨基酸大环偶联物最终在多个动物模型中都表现出优异的抗肿瘤活性,其抑制肿瘤生长效果明显优于实验对照组(包括野生型干扰素,PEG偶联物和线性聚氨基酸偶联物)。该研究较为深入地探讨了蛋白质-聚氨基酸大环偶联物的活体药学表现,为人们设计下一代蛋白质/多肽类药物提供了新的思路。论文所报道的干扰素-聚氨基酸大环偶联物表现出广阔的应用前景和可观的转化潜力,预期适应症包括卵巢癌、前列腺癌、皮肤淋巴瘤和黑色素瘤等多种恶性肿瘤,同时也有一定的潜力用于乙肝和丙肝等病毒性疾病。该工作近期在线发表于Journal of the American Chemical Society (http://pubs.acs.org/doi/10.1021/jacs.7b13017)。

图2. 干扰素-聚氨基酸大环偶联物具有肿瘤滞留多、组织渗透深和抑瘤活性好等特点。
北京大学化学院博士生侯颖钦和联合培养硕士生周雨为该论文共同第一作者,吕华研究员为论文通讯作者。该工作获得了国家重点研发计划纳米专项(2016YFA0201400)和国家自然科学基金委(21722401, 21474004, 和 21434008)的支持。

 北大化学微信
北大化学微信