![]()

With genome sequencing completed for many species on earth, we are left with a daunting number of predicted protein products whose functions remain to be deciphered. The function of a protein is precisely realized and tightly regulated with both temporal and spatial resolution in cell, mainly through action on its catalytic center, interaction with small molecule ligand as well as by post-translational modification (PTM) on certain residue sidechains. Discovery of these “functional sites” will be very informative and valuable for us to understand the important role that each protein plays in complicated biological processes.
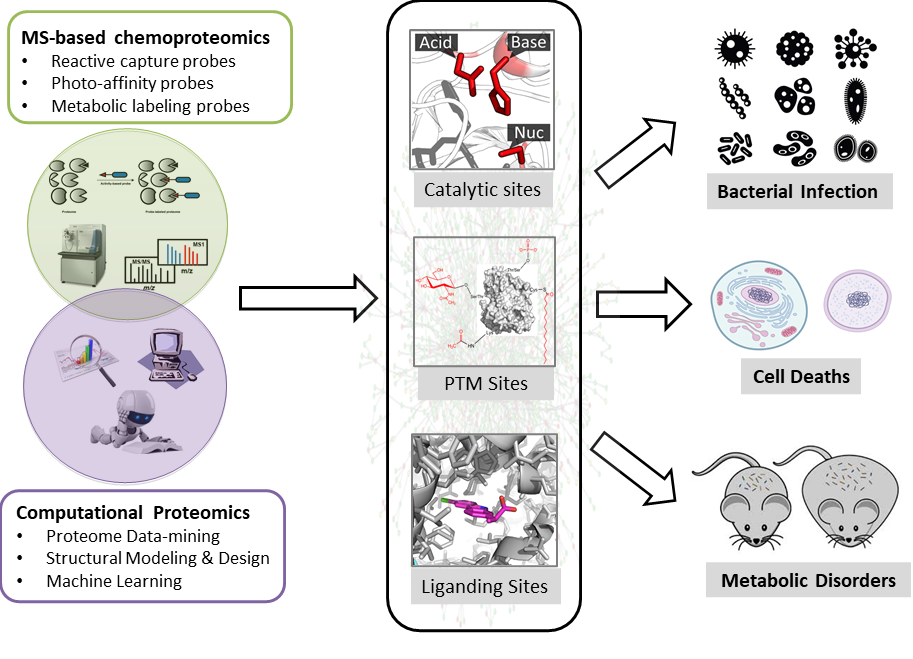
Our research programs focus on developing cutting-edge chemical biology and proteomics tools to discover and interrogate functional sites in proteomes by a combination of experimental and computational approaches (Figure 1). Experimentally, we furnished the 'Activity-Based Protein Profiling (ABPP)' technology pioneered by Cravatt and colleagues with new chemical probes and new analytical methods to quantitatively mine functional proteomes with site-specific resolution. Computationally, we developed and employed tools of bioinformatics, structural modeling and design, and artificial intelligence to systematically discover and predict novel functional sites in proteomes. We are generally interested in three major areas:
These studies have the great potential to provide penetrating mechanistic insights into the molecular basis for numerous diseases functionally linked to metabolic disorder as well as to integrate and streamline efforts in inhibitor discovery, drug design and the functional annotation of uncharacterized enzymes in the post-genomic era.
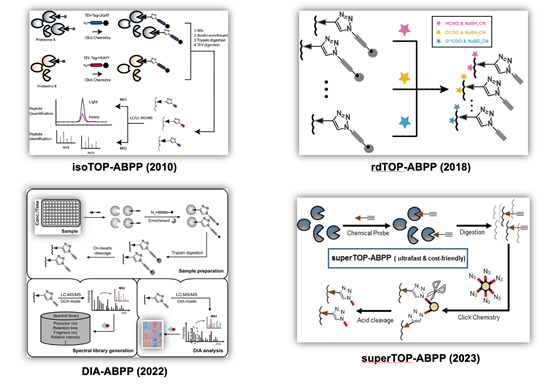
Cysteines, as one of the most intrinsically nucleophilic amino acids, play important roles in proteins involved in diverse biological processes. In addition to acting as catalytic residues in many enzymes, cysteines are also subjected to covalent modifications by endogenous reactive metabolites and clinical drugs. Discovery and characterization of functional cysteines will help to annotate their protein functions as well to identify novel drug targets. Previously, a chemical proteomic platform, named as “isoTOP-ABPP” (Figure 2, top left), was developed by Cravatt and colleagues to experimentally profile cysteines with intrinsically heightened reactivity in human proteomes and the quantitative analysis showed a strong correlation between the chemical reactivity of a cysteine and its functionality (Nature 2010, 468, 790-795) and a “competitive isoTOP-ABPP” platform was also developed that enables quantitative and site-specific profiling of targets of cysteine-reactive electrophilic metabolites (Nat. Methods 2014, 11, 79-85).
Our research group has a continuing interest in advancing the isoTOP-ABPP technology for functional cysteinome profiling with expanded proteome coverage, enhanced quantification multiplexity and improved usability. We first revised isoTOP-ABPP with a more convenient and economic labeling strategy based on the reductive dimethylation and the resulting “rdTOP-ABPP” method (Figure 2, top right) not only enables triplex quantitation, but also works seamlessly with any type of cleavable enrichment tags, many of which are commercially available (Anal. Chem. 2018, 90, 9576-9582). We next combined the data-independent acquisition (DIA) mass spectrometry with ABPP to develop an efficient label-free quantitative chemical proteomic method, DIA-ABPP (Figure 2, bottom left), with good reproducibility and high accuracy for multiplex quantification. The power of DIA-ABPP for comprehensive profiling of functional cysteineome was demonstrated in three distinct applications, including dose-dependent quantification of cysteines’ sensitivity toward a reactive metabolite, screening of ligandable cysteines with a covalent fragment library, and profiling of cysteinome fluctuation in circadian clock cycles (J. Am. Chem. Soc. 2022, 144 (2), 901-911; Curr. Res. Chem. Biol. 2022, 2, 100024.). More recently, we developed a simplified and ultrafast “superTOP-ABPP” pipeline (Figure 2, bottom right), which cuts down the averaged sample preparation time and significantly improves the sensitivity and coverage of site-specific cysteinome profiling (J. Proteome Res. 2023, DOI: 10.1021/acs.jproteome.3c00179). The rich chemoproteomic data obtained were also fed to machine learning algorithms to enable computational prediction and discovery of reactive cysteines in other organisms and species (Biochemistry 2018, 57 (4), 451-460). By applying these methods, we have identified functional cysteines in specific biological systems such as ferroptosis (Cell Death Differ. 2023, 30 (1), 125-136.) as well as cysteine targets of clinical drugs (Mol. Pharm. 2018, 15, 2413-2422; RSC Chem. Biol. 2023, 4 (9), 670-674.).
Selenium (Se), as an essential trace element, plays crucial roles in many organisms including humans. The biological functions of selenium are mainly mediated by selenoproteins, a unique class of selenium-containing proteins in which selenium is inserted in the form of selenocysteine (SeCys). SeCys is considered as the 21st proteinogenic amino acid and although structurally similar to cysteine, it has much higher chemical reactivity that the sulfur counterpart. Due to their low abundance and uneven tissue distribution, detection of selenoproteins within proteomes is very challenging, and therefore functional studies of these proteins are limited. To address this challenge, we developed a computational method, named as selenium-encoded isotopic signature targeted profiling (SESTAR), which utilizes the distinct natural isotopic distribution of selenium to assist detection of trace selenium-containing signals from shotgun-proteomic data. SESTAR can greatly enhance the detection sensitivity of native selenoproteins from tissue proteomes in a targeted profiling mode (ACS Cent. Sci. 2018, 4 (8), 960-970; Methods Enzymol. 2022, 662, 241-258). Taking advantage of the unique isotopic signature, we recently developed a chemical proteomic strategy named "SElenoprotein Turnover Rate by Isotope Perturbation (SETRIP)" to quantitatively monitor the turnover dynamics of selenoproteins at the proteomic level (Anal. Chem. 2022, 94 (27), 9636-9647.).
Protein post-translational modifications (PTMs) are covalent moieties introduced to the amino acid side chains or termini of proteins, either enzymatically or chemically, which can change the physicochemical properties of target proteins, and lead to structural changes, localizations, activities and binding partners. To date, many different types of PTMs in prokaryotic and eukaryotic cells have been identified. PTMs are usually sub-stoichiometric with low abundance, heterogeneous in their structures, and dynamically regulated, which make them difficult to identify and analyze.
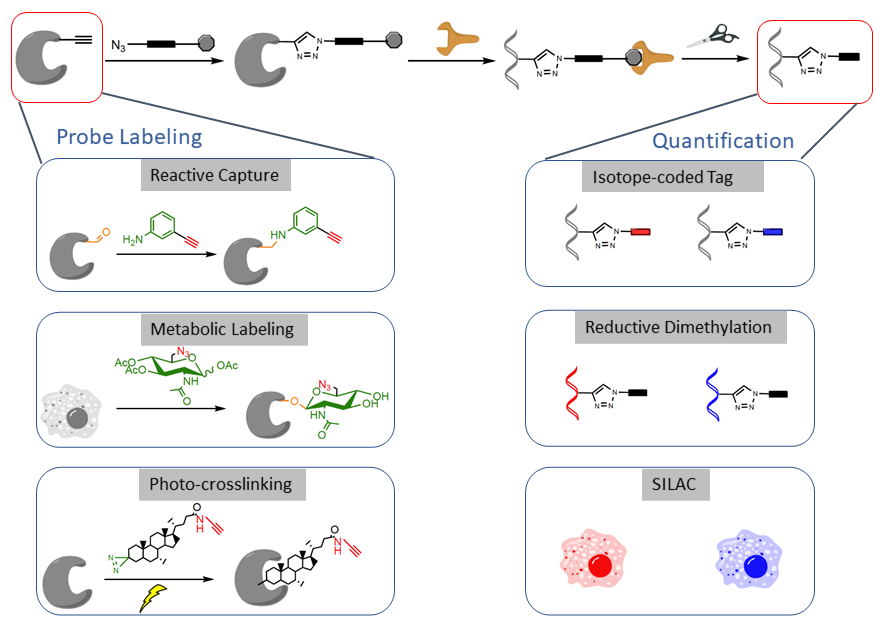
We are interested in developing chemical probes to covalently label the PTM of interest and enrich them for site-specific quantitative proteomic analysis (Figure 3). Such a ‘‘chemical proteomic’’ or ‘‘chemoproteomic’’ strategy can be generally classified into three approaches depending on the chemical probes used: (1) ‘‘reactive capture’’ by chemoselective probes; (2) select labeling of reactive amino acids in proteomes; and (3) metabolic or direct labeling by bioorthogonal PTM analogue probes. After probe labeling, the samples can be subjected to analysis by quantitative proteomic techniques such as isoTOP-ABPP, reductive dimethylation, SILAC or TMT or SILAC.
By applying the above-mentioned approaches, we have successfully developed chemical proteomics strategies for profiling an array for functional PTMs, both enzymatical and non-enzymatical, including S-carbonylation (Redox Biol. 2017, 12, 712-718; J. Am. Chem. Soc. 2018, 140 (13), 4712-4720; Chem. Res. Toxicol. 2019, 32 (3), 467-473.), S-itaconation (Nat. Chem. Biol. 2019, 15 (10), 983-991; J. Am. Chem. Soc. 2020, 142 (25), 10894-10898; Chem. Sci. 2021, 12 (17), 6059-6063.), O-GlcNAcylation (Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (33), E6749-E6758; Angew. Chem. Int. Ed. Engl. 2018, 57 (7), 1817-1820; ACS Chem. Biol. 2018, 13 (8), 1983-1989.), O-Phosphopantetheinylation (Angew. Chem. Int. Ed. Engl. 2020, 59 (37), 16069-16075; Chembiochem 2021, 22 (8), 1357-1367.), N-homocysteinylation (Chem. Sci. 2018, 9 (10), 2826-2830; Chem. Commun. 2019, 55 (25), 3654-3657.), N-lipoylation (J. Am. Chem. Soc. 2022, 144 (23), 10320-10329.), and glyoxal-derived modifications (Future Med. Chem. 2019, 11 (23), 2979-2987; ACS Chem. Biol. 2022, 17 (8), 2010-2017.). The performance and sensitivity of such methods can be further enhanced with the aid of novel and sophisticated computational algorithms to uncover novel PTM sites in proteomes, such as N-itaconylation (J. Am. Chem. Soc. 2023, 145 (23), 12673-12681) and N-lactylation (Sci. Adv. 2023, 9 (14), eadf1416.).
In recent years, the high-throughput screening of biologically active small molecules using chemical genetics has been one of the frontier topics in the field of chemical biology. However, these large-scale screening efforts ultimately face the challenge of answering the important scientific question: what are the cellular targets of and mechanisms of action for these bioactive molecules? In addition to exgenously screened ligands, a large array of biologically active endogenous metabolites also exert diversified functions within the organism by non-covalent binding to, thereby regulating or altering the structure and function of these proteins. It is therefore of great importance to determinine their target proteins of these bioactive ligands in order to unveil the molecular basis of their biological activity.
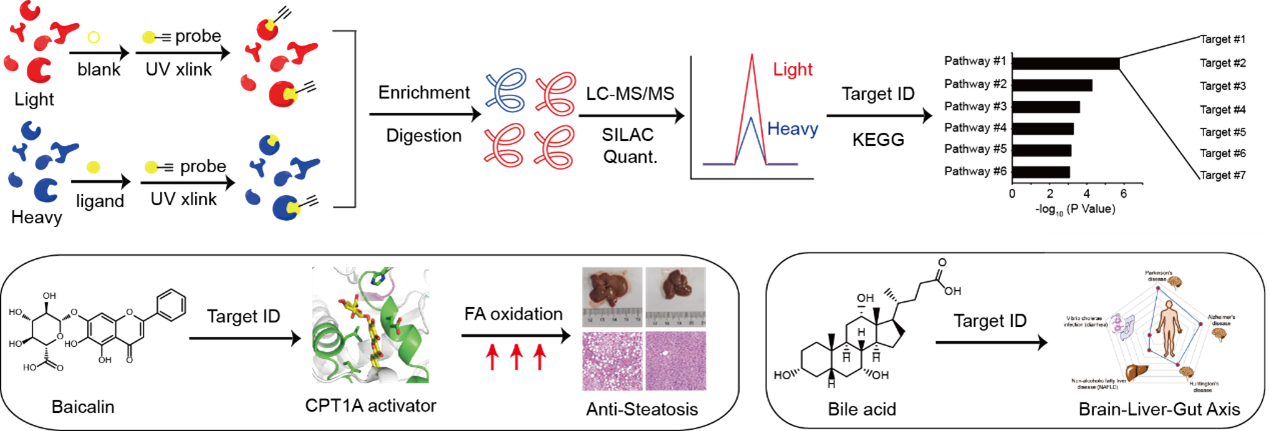
Our researh group is interested in developing and applying quantitative chemoproteomic methods to deconvolute targets of bioactive ligands at the whole-cell proteome level. The approaches can be either based on photo-affinity labeling using multifunctional analouge chemical probes or by using the native ligands in the pipneline of thermal proteome profiling (TPP) and competitive isoTOP-ABPP, and the bioactive ligands could be either natural products (Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (26), E5896-E5905.), endogenous metabolites (ACS Cent. Sci. 2017, 3 (5), 501-509; ACS Chem. Biol. 2022, 17 (9), 2461-2470.), clinically approved drugs (Mol. Pharm. 2018, 15 (6), 2413-2422; RSC Chem. Biol. 2023, 4 (9), 670-674.) or synthetic ligands (Chembiochem 2021, 22 (1), 129-133; Angew. Chem. Int. Ed. Engl. 2020, 59 (45), 20147-20153.).
For example, to decipher the mechanism of action of baicalin, a natural flavonoid compound isolated from a Chinese herbal medicine reported with intriguing anti-steatosis activity, we synthesized a photo-affinity probe of baicalin and performed quantitative chemical proteomic profiling to identify a list of baicalin-bound proteins in cells (Figure 4, left). Guided by further bioinformatic analysis and genetic screening, we focused on the interaction between baicalin and carnitine palmitoyl-transferase 1A (CPT1A), a crucial enzyme in control of fatty acid β-oxidation (FAO) in hepatocytes. Biochemical assays and computational docking experiments revealed that baicalin directly binds to a defined pocket on CPT1A and allosterically activates the enzyme to accelerate fatty acid degradation. In vivo studies confirmed that baicalin can significantly ameliorate diet-induced hepatic steatosis and obesity, as well as associated metabolic disorders in mice. Notably, the beneficial effect of baicalin critically depends on its interaction with CPT1 as mice with a baicalin-insensitive mutant of CPT1A completely lost response to the baicalin treatment (Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E5896-E5905).
In addition to bioactive natural products, we also developed a chemoproteomic strategy to profiling binding proteins of bile acids (BAs), a class of endogenous metabolites that act as signaling molecules to regulate lipid and glucose metabolism as well as gut microbiota composition in the host (Figure 4, right). By combining multiple BA-based photoaffinity probes with a SILAC-based quantitative proteomic approach, we identified >600 BA-interacting protein targets including known endogenous receptors and transporters of BA, and biochemically verified BA’s interaction with some novel protein targets. Bioinformatic analysis revealed that these newly identified BA-interacting proteins are mainly enriched in functional pathways predicted with strong implications in neurodegenerative diseases, non-alcoholic fatty liver disease and diarrhea (Zhuang, S. et al, ACS Cent. Sci. 2017, 3, 501-509). More recently, we also performed the profiling in bacterial proteomes and found a novel sensor that binds with BA and activates a downstream signaling pathway to help efflux of BA from bacteria, resulting in BA tolerance (ACS Chem. Biol. 2022, 17 (9), 2461-2470.).
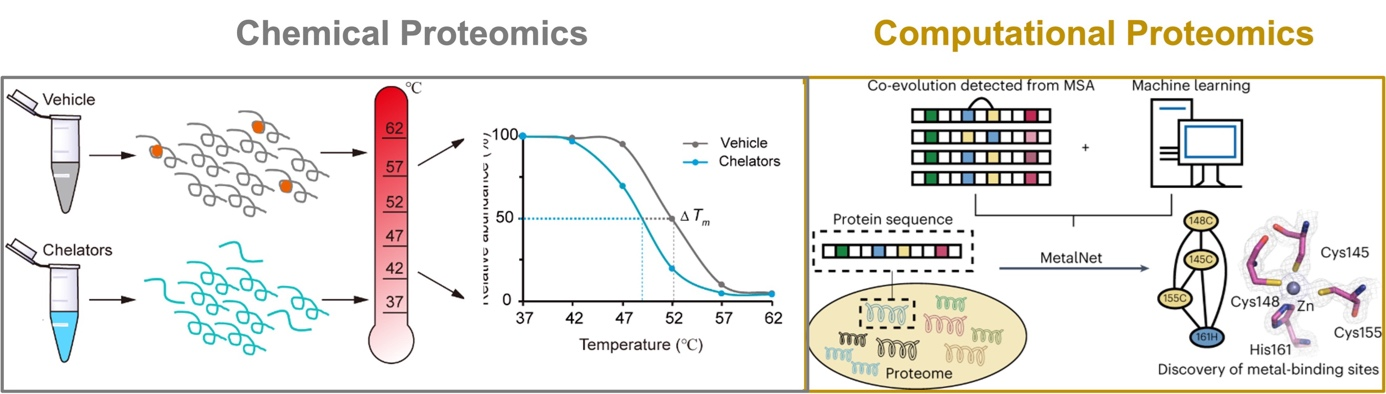
Metal-binding proteins (MBPs) are universally distributed in all biological systems, which are estimated to constitute about one third of the entire proteome. They play indispensable roles in various biological processes, including catalysis, structural stabilization and signal transduction. It has also been reported that many human diseases are closely related to dysfunctional MBPs, such as neurodegenerative diseases and cancers. Traditional photo-affinity chemoproteomic technologies are not applicable to metalloproteomes and current bioinformatic methods usually predict MBPs based on known consensus sequence and/or structural motifs. To provide alternative and complementary approaches for mining metalloproteomes, we have explored two directions. Experimentally, we developed a chemoproteomic method named METAL-TPP for global discovery of MBPs in proteomes, which operates by extracting metals from MBPs with chelators and logging the resulting structural perturbation of MBPs with thermal proteome profiling (Nat. Chem. Biol. 2023, accepted in principle). Computationally, we developed a co-evolution-based pipeline named 'MetalNet' to systematically predict metal-binding sites in proteomes (Nat. Chem. Biol. 2023, 19 (5), 548-555.). By applying both methods, we identified a large number of potential MBPs, which substantially expands the currently annotated metalloproteomes. We also biochemically and structurally validated some previously unannotated metal-binding sites in several proteins. Metal-TPP and MetalNet provide unique and enabling tools for interrogating the hidden metalloproteome and studying metal biology.