Allostery plays important roles in many biological processes, such as enzyme catalysis and signal transduction. Allosteric drugs have raised much attention due to their high specificity and possibility of overcoming existing drug-resistant mutations. However, optimization of allosteric molecules faces great challenges as allosteric molecules usually have flat structure-activity relationships and higher binding affinity does not always correspond to better activity. In an allosteric site, the contribution of each residue to the allosteric effect is different, among which the key residues determine the direction and strength of allosteric signal transduction, while the other residues mainly play an auxiliary role in binding. It is of great significance to identify residues that trigger allosteric signaling, which are referred as key allosteric residues or key allo-residues. Currently there is no effective method to predict key allosteric residues.
The teams led by Prof. Luhua Lai at the College of Chemistry and Molecular Engineering, and Center for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, in collaboration with Prof. Minghua Deng at the School of Mathematical Sciences, and Center for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, and Prof. Xiaolei Zhu at the School of Sciences, Anhui Agricultural University, developed the first systematic computational method, KeyAlloSite to predict key allosteric residues. They demonstrated that orthosteric and allosteric sites are coupled in protein function evolution, and weak coevolutionary couplings contain important information of protein allosteric regulation function. KeyAlloSite provides a powerful tool for designing and optimizing allosteric drugs and designing functional proteins. This work was published online by the journal eLife on February 17, 2023 (https://elifesciences.org/articles/81850).
During evolution, functionally coupled residues in proteins coevolve, and the evolutionary coupling pattern between residues can be estimated from multiple sequence alignment (MSA). Since the perturbation at an allosteric site can affect the function of the orthosteric site, they analyzed the evolutionary coupling strength (ECS) between the orthosteric and allosteric pocket, as well as all the other pockets. They found that orthosteric and allosteric pocket are more evolutionarily coupled to each other than the orthosteric and other non-functional pockets.
They further performed pairwise comparison of the differences in the evolutionary coupling scores of residues in the allosteric pocket, and calculated the number of significant differences of each residue in the allosteric pocket and normalized to Z-scores. Residues were predicted as key allo-residues if their corresponding Z-scores were greater than the threshold (Fig. 1). The key allo-residues predicted by KeyAlloSite are in accordance with experimental results in several protein systems. For example, in the tyrosine-protein kinase ABL1 (BCR-ABL1), the key allo-residue L359 predicted by KeyAlloSite plays a key role in allosteric signaling. Since there is no favorable interaction between the allosteric ligand hit 4 and L359, hit 4 has no inhibition activity despite its relatively high binding affinity. The favorable hydrophobic interactions between hit 5 and L359 makes it inhibit the kinase activity albeit with a weaker binding affinity compared to hit 4 (Fig. 2).
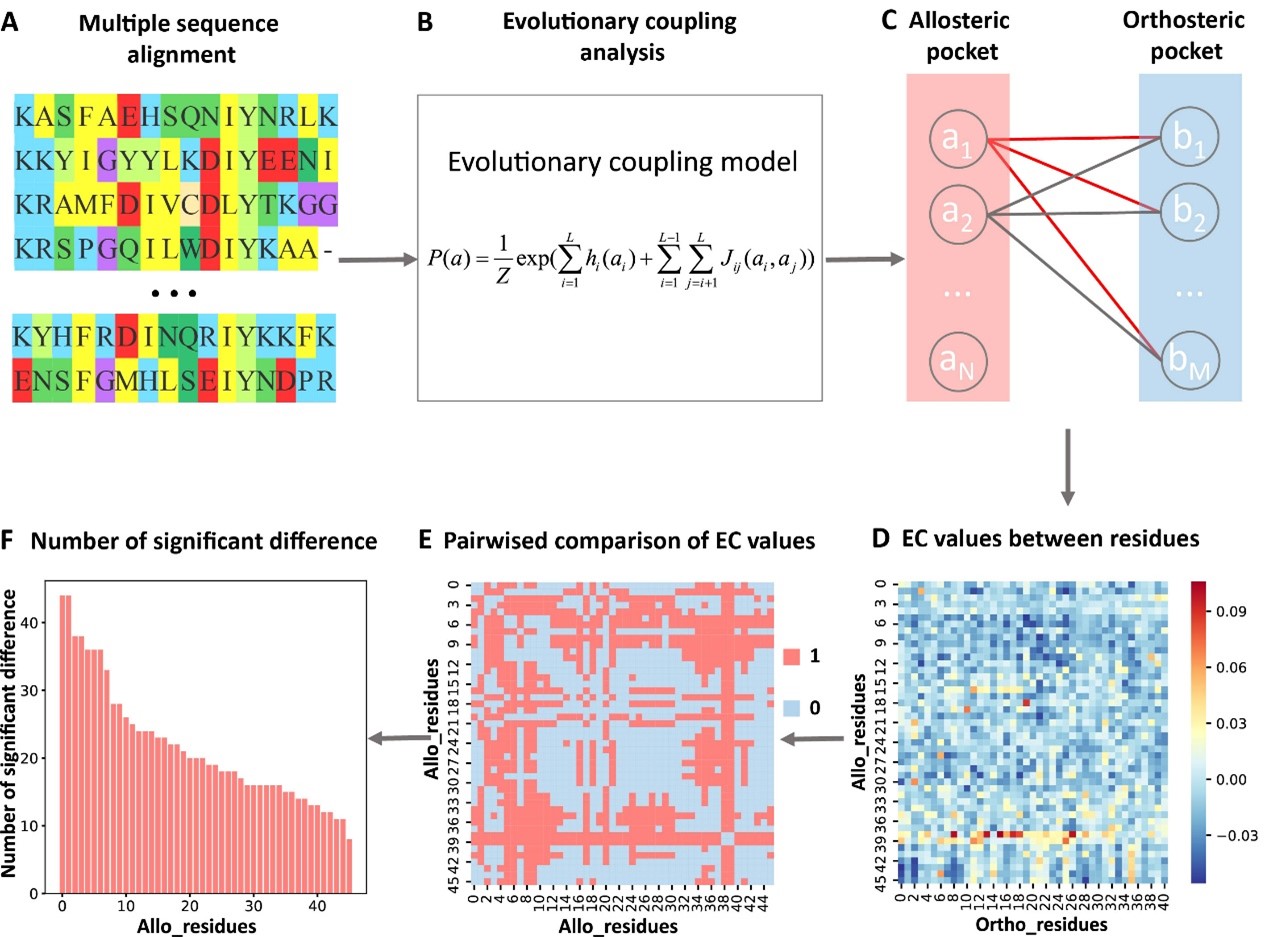
Fig.1 Steps to identify key allo-residues.
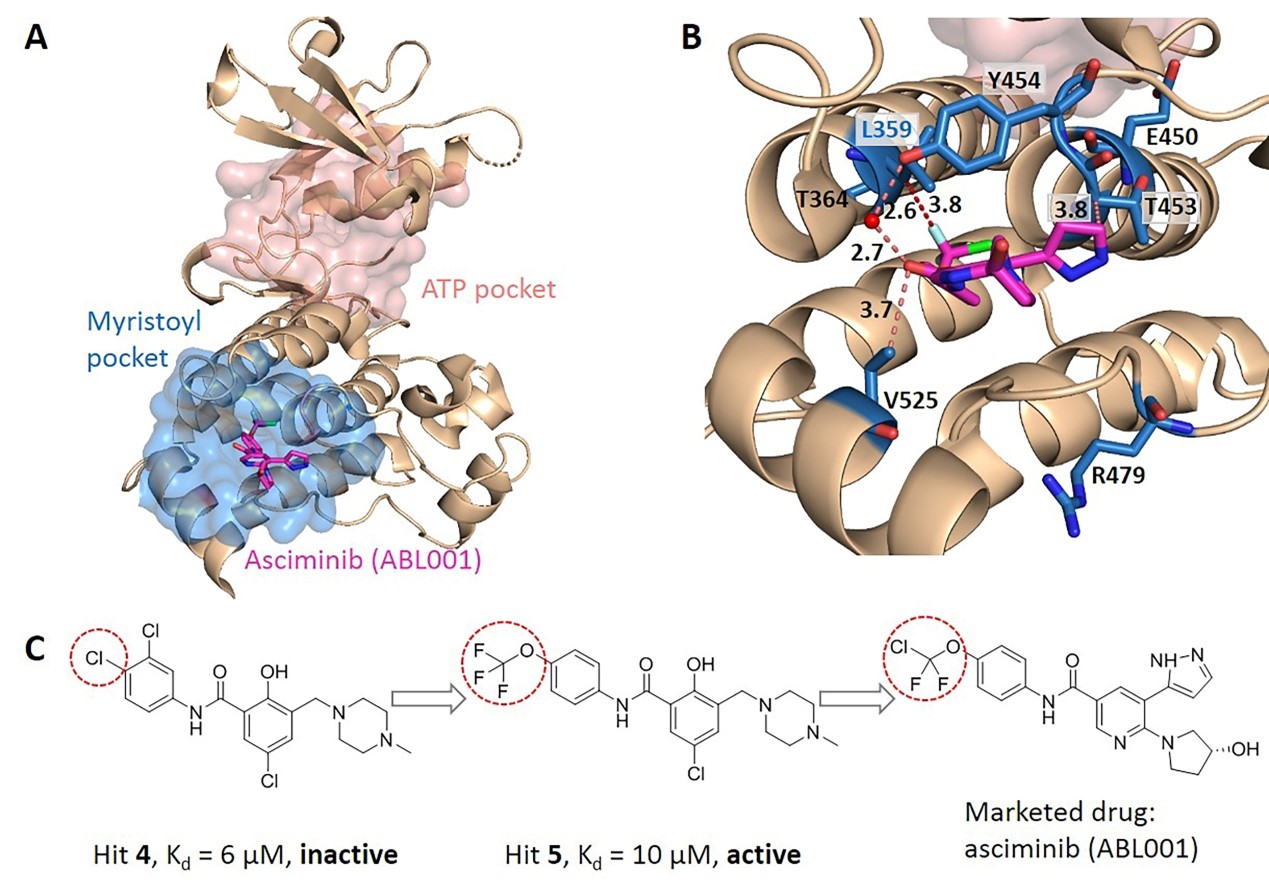
Fig. 2 Key allo-residues predicted by KeyAlloSite in BCR-ABL1.
KeyAlloSite can also be used to predict key allosteric residues distant from the catalytic site that are important for enzyme catalysis. For example, in Candida antarctica lipase B (CALB), the key allo-residues A225 and V37 predicted by KeyAlloSite are important for the catalytic activity of the enzyme. A225M improves the catalytic efficiency of the enzyme by about 11 folds, and V37I improves the catalytic efficiency of the enzyme by about 3 folds.
Dr. Juan Xie, a postdoctoral researcher at the Center for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, is the first author of the paper. Prof. Luhua Lai at the College of Chemistry and Molecular Engineering, and Center for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University is the corresponding author. Prof. Minghua Deng at the School of Mathematical Sciences, and Center for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, and Prof. Xiaolei Zhu at the School of Sciences, Anhui Agricultural University, provided key guidance and assistance for this research. Dr. Weilin Zhang, a former postdoctoral researcher of the Lai group contributed to this work. This research was supported by the National Natural Science Foundation of China, the Chinese Academy of Medical Sciences, the Beijing National Laboratory for Molecular Sciences, and the Peking-Tsinghua Center for Life Sciences.
Original link for the paper: https://elifesciences.org/articles/81850.