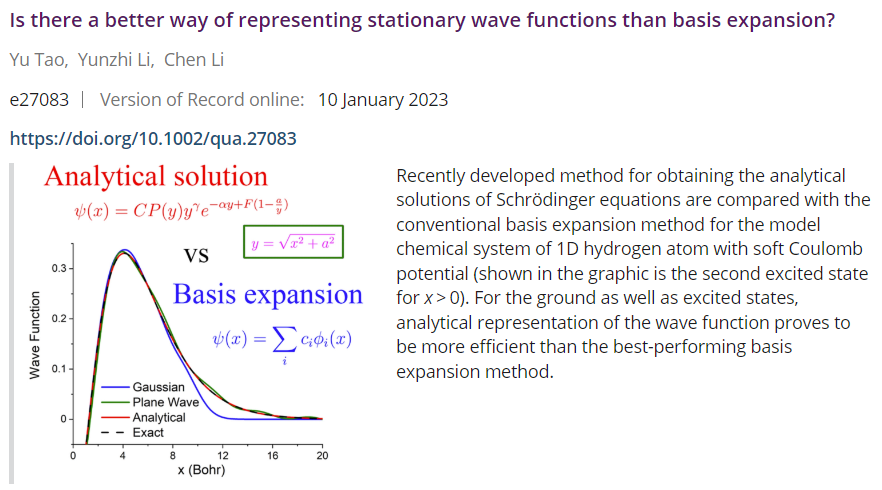
Basis sets are widely used in quantum chemistry calculations. The present-day routinely used basis sets are based on the contracted Gaussian functions (CGFs), by which one transforms the computationally expensive numerical Coulomb integrations into a weighted sum of analytical Gaussian expressions. With some compromise of accuracy (cusp condition, etc.), it achieves a lower computational cost.
Yet, a natural and crucial question is: is there a better way of representing stationary wave functions than basis expansion? In this work, we show that there exists. Using a similar strategy to our previous work, by utilizing the analytical structure of the exact wave functions, we factorize them into a polynomial prefactor encoding the nodal information, a non-integer power prefactor, and an exponentially decaying term modulated by a modulator function.
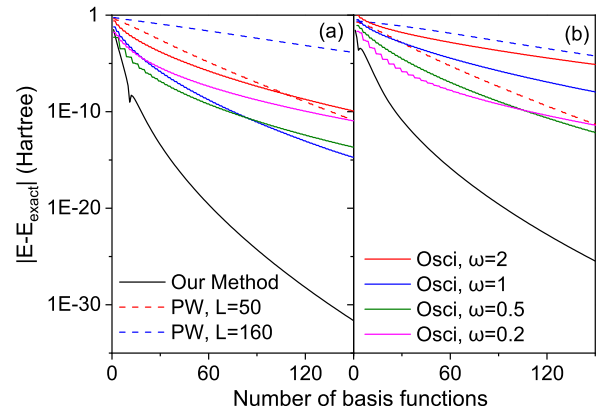
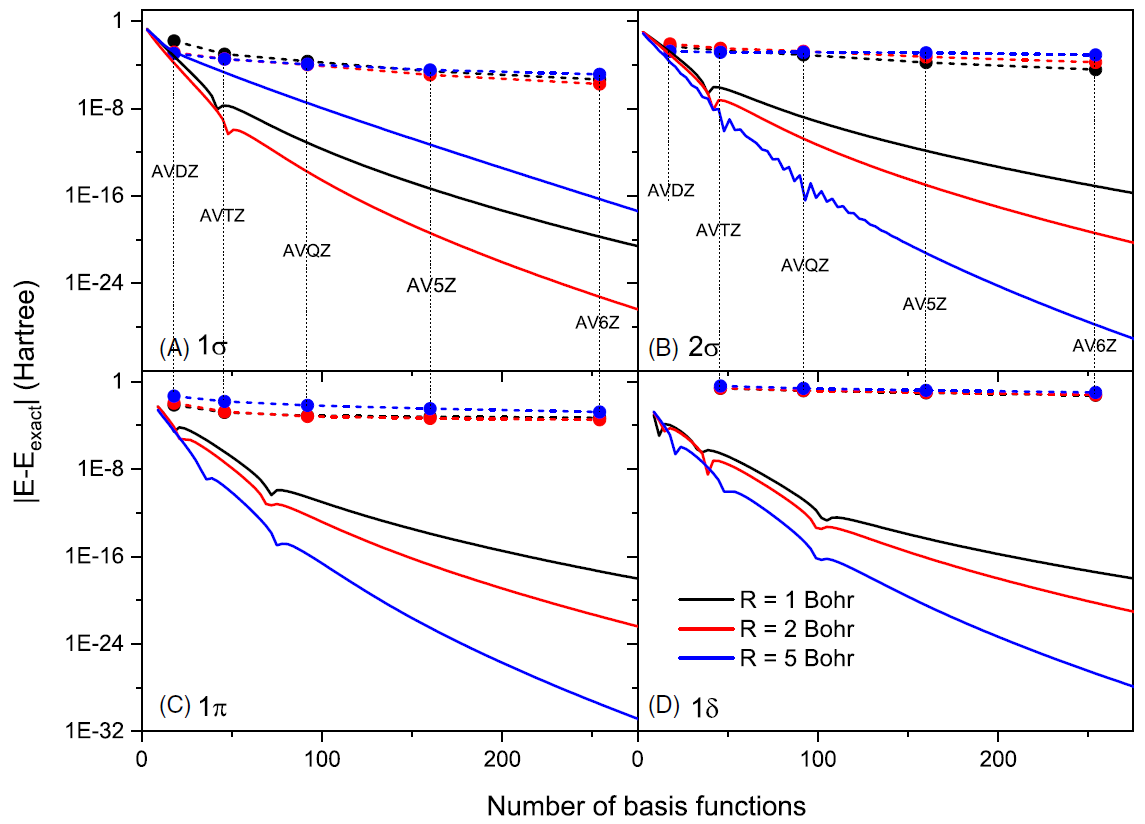
Our numerical tests in 1D H atom revealed the great advantage of our methods over basis expansion methods. In the comparison, both local and nonlocal basis sets have been considered, where all of them converge much more slowly than our method. Similar tests on 1D H2+ molecular ion show similar results. Furthermore, by performing the real-world 3D H2+ calculation as a validation, we show that our 1D conclusion is transferable to 3D.
The physics of the modulator function F(z) is also intriguing. Although it is formally defined by summing over infinitely many small terms, it is packed up as an O(1) term in the entire space and is thus less crucial than the other factors. This also explains the successes of the variational attempts in the literature: by a not too bad approximation to F, one can usually obtain reasonably good results.
The exact analytical formulas derived in this work also shed light on the plausible ways of approximations, to which approximating F(z) could be a promising approach. Although we have only considered single-electron systems in the present work to demonstrate the superiority of our method over basis expansion, this suffices for a proof of concept. It would be even more inspiring to extend our exact analytical formula to many-electron systems and demonstrate its computational advantage; and our preliminary results seem to indicate that this advantage should be even larger for many-electrons.
Yu Tao is the first author of the paper and Yunzhi Li also made significant contributions. Congratulations to both of them.
For more details, see the following links: https://onlinelibrary.wiley.com/doi/10.1002/qua.27083
