
Molecular orbital (MO) is a widely used concept in chemistry and the key for studying electronic structure of molecules. In fact, almost all modern ab-initio methods are built upon or closely related to MOs; a few examples include the Hartree-Fock theory, Kohn-Sham density functional theory, M∅ller-Plesset perturbation theory and so on. The simplest MOs are defined as the solutions to the single-electron Schrödinger equation of the simplest molecule H2+. The resulting orbitals and energies as functions of the nuclear separation R have been used as a textbook problem for understanding the simplest chemical bond. However, unlike atomic orbitals (AOs) whose exact analytical formulas have been completely clarified, exact expressions for MOs are regarded as too complicated to be accessible.
Efforts on finding the exact analytical formulas of MOs of H2+ date back to 1927-1931, right after quantum mechanics was established. Important progress was made by Hylleraas et al in separating variables in spheroidal coordinates (also called confocal elliptic coordinates). However, the remaining problem is still difficult; one cannot easily write down elegant formulas resembling the AOs of hydrogen. This problem kept attracting attentions of theoretical physicists and chemists until 1960s, yet a complete analytical form remains elusive. After the 1980s until recently, efforts have been focused on numerically approximating energies of arbitrary excited states of H2+, given the limitation of the standard method of linear combinations of atomic orbitals (LCAO) for these states.
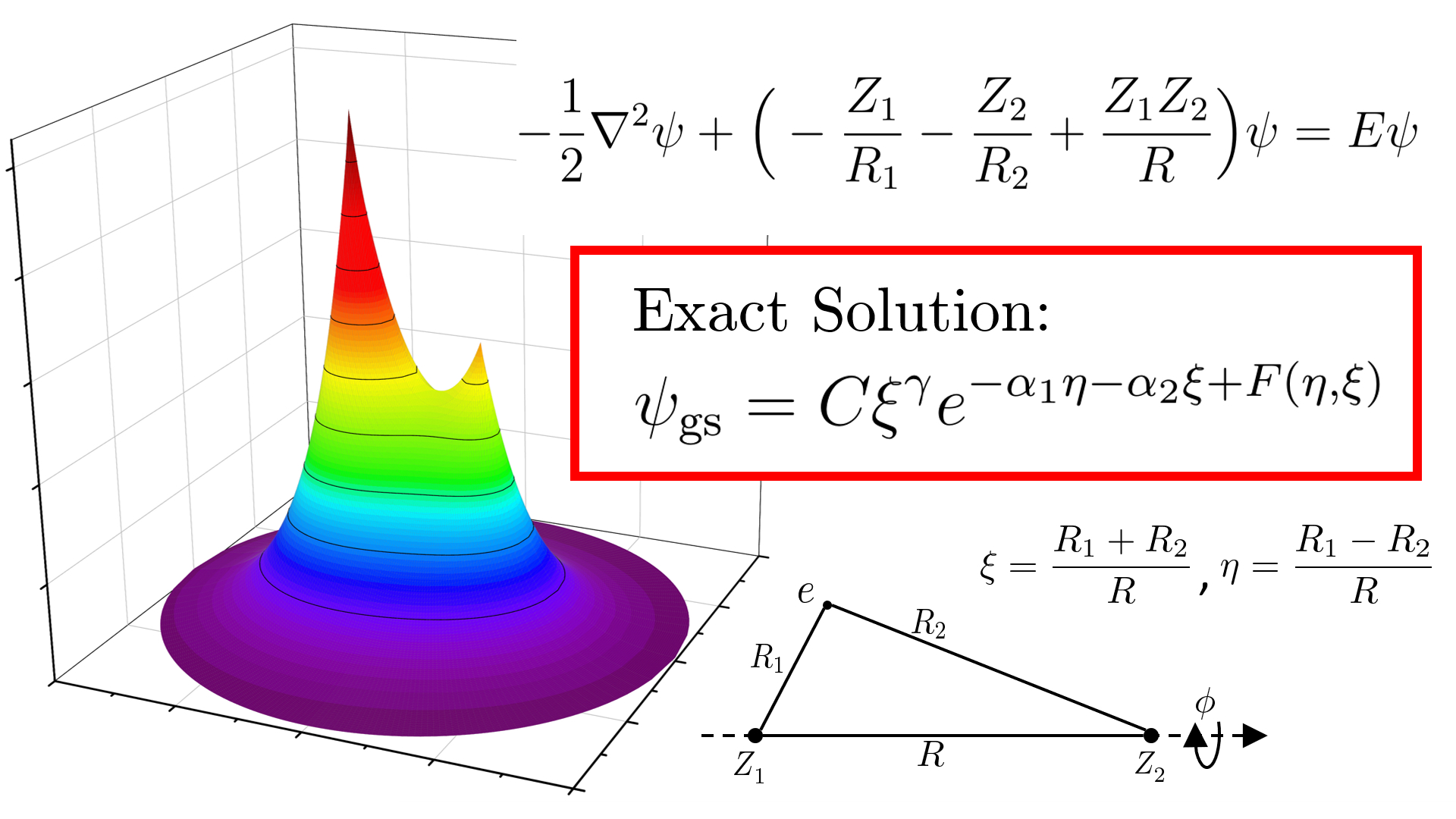
In this paper, by using our recently developed method for solving Schrödinger equations, we completely solve this longstanding problem by writing explicitly the exact analytical form of MOs for a diatomic molecule with arbitrary nuclear charges, covering the special case of H2+. Our result is in a completely factorized form, which we claim to be the best representation. For the ground state, our factorization includes a power prefactor, an exponentially decaying term, and a modulator function on the exponential; while for excited states it involves additional factors accounting for nodal surfaces and the magnetic quantum number. Through our formula we have achieved a unification in the analytical forms of AOs and MOs and have revealed the general structure of single-electron wave functions for Coulomb systems. In particular, one can readily find that the modulator function of AOs vanishes because of spherical symmetry.
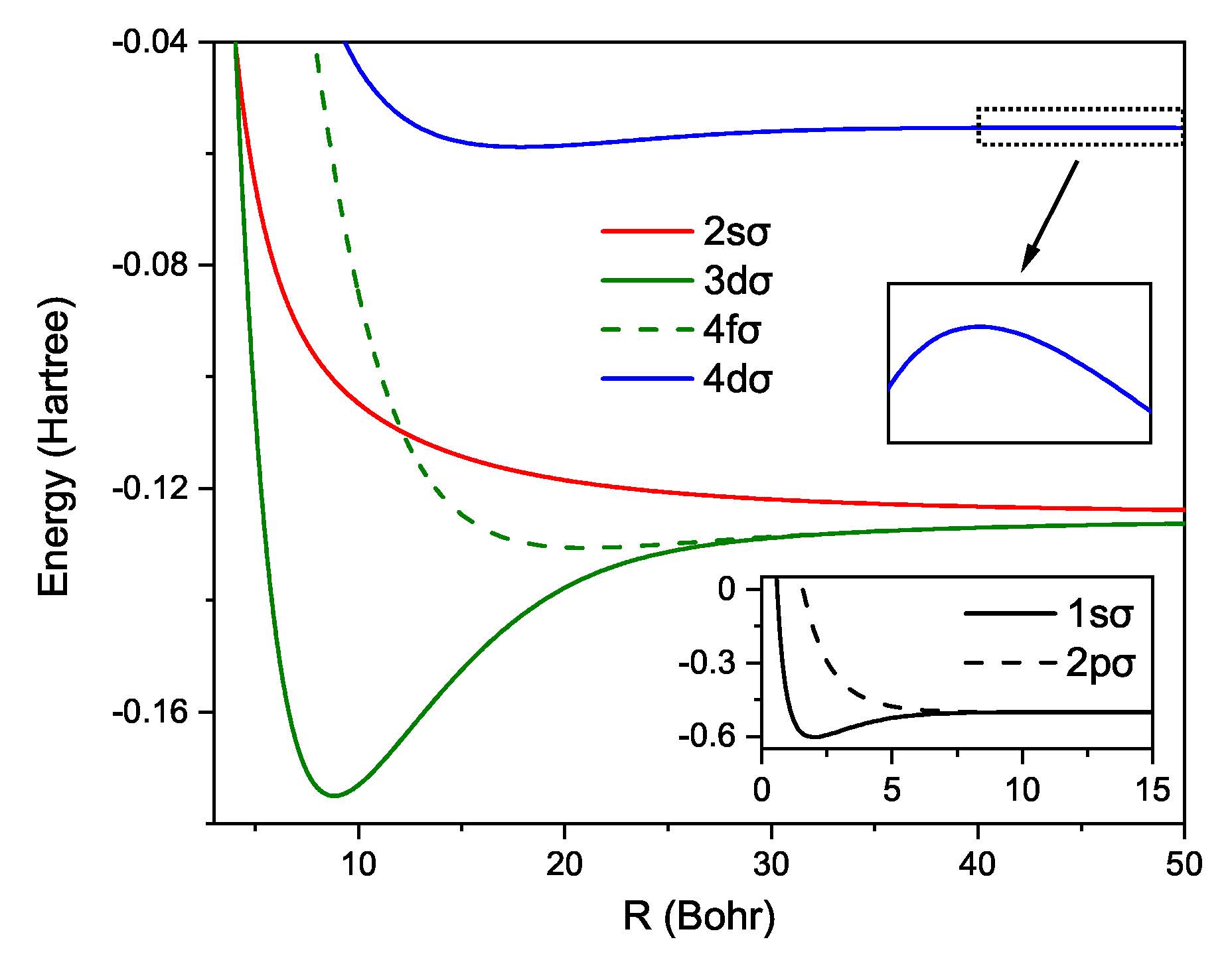
The usefulness of our formulas has been demonstrated through several examples, to name a few: (i) our formula gives immediate insight into possible accurate variational approximations; (ii) the modulator function proposed in this work is potentially useful for constructing density functional approximations to address the delocalization error; (iii) importantly, we have observed unexpected extreme points along the exact potential energy curves, which calls our attention to the limitations of the well-known concepts of bonding and antibonding.
The exact analytical formula of diatomic MOs obtained in this work is a major breakthrough in this very challenging and fundamental problem, and shall give us great insight into the nature of chemical bond. Because of its potential for broad public interest, our paper has been selected to be featured as an ACS Editors' Choice in addition to being published in ACS Omega. Yunzhi Li is the first author of this paper. Congratulations to Yunzhi!
For more details, see the following links: https://pubs.acs.org/doi/10.1021/acsomega.2c01905
